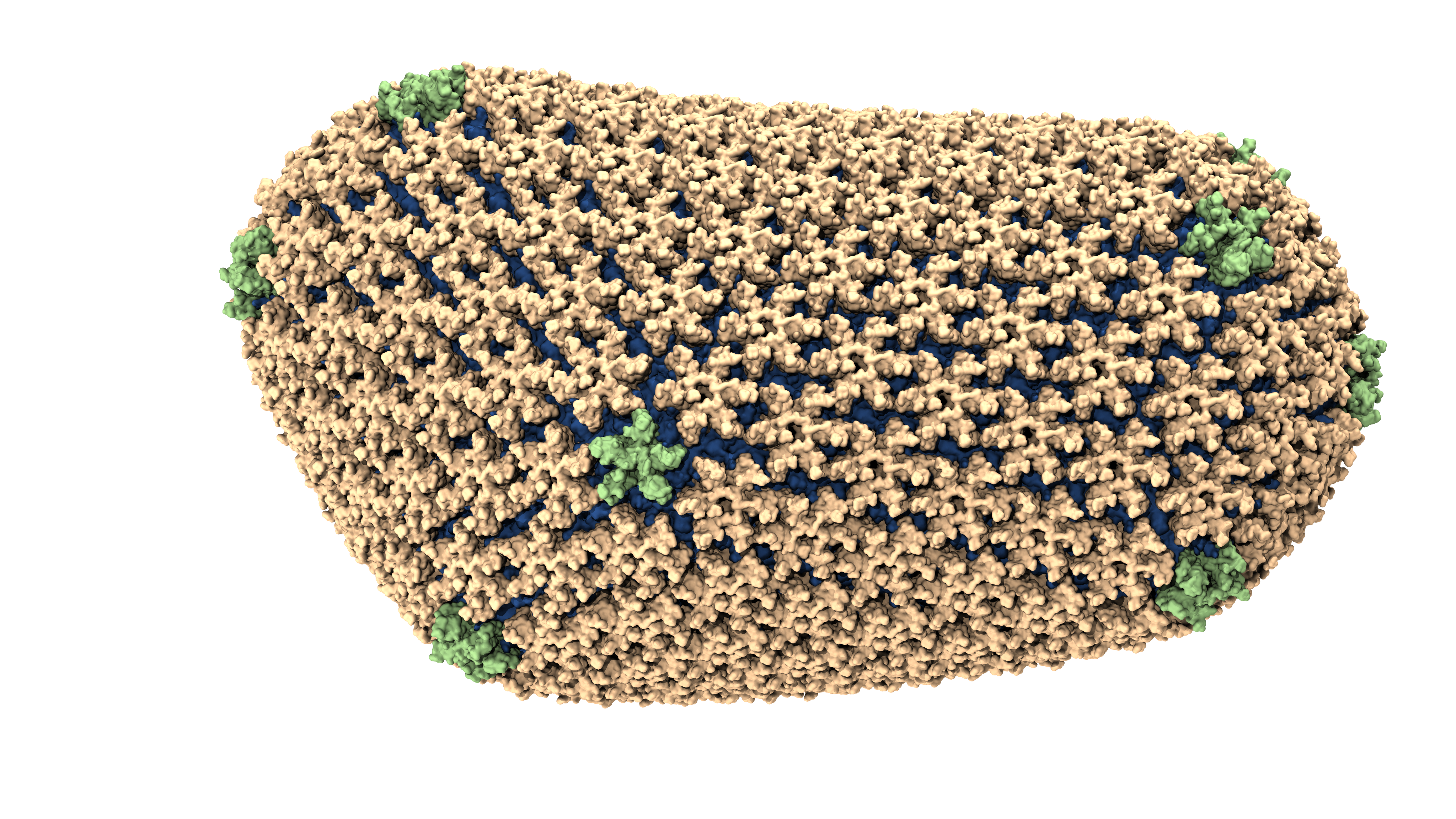
An atomic model of the HIV-1 capsid, or shell.
HIV/AIDS has caused an estimated 36 million deaths, according to the World Health Organization, and remains a major menace worldwide. Today approximately 35 million people are living with the human immunodeficiency virus (HIV), including more than 1 million individuals in the United States.
The tendency of HIV to mutate and resist drugs has made it particularly difficult to eradicate. Some treatments have shown progress in slowing or even stopping the progress of the virus, but no cure or vaccine has been discovered that can truly stamp out the disease.
In the last decade scientists have begun using a new weapon in the fight against HIV: supercomputers.
Scientists harness the power of thousands of computer processors simultaneously to better understand how the HIV virus interacts with the cells it infects, to discover or design new drugs that can attack the virus at its weak spots and even to use genetic information about the exact variants of the virus to develop patient-specific treatments.
Among the researchers using supercomputers to study HIV is Klaus Schulten, the keynote speaker at the 2014 International Supercomputing Conference in Leipzig, Germany. Schulten, a professor of physics at the University of Illinois at Urbana-Champaign, invented the Nanoscale Molecular Dynamics (NAMD) software program, one of the most widely used tools for understanding diseases at a molecular level.
Supported by the National Science Foundation and using some of the nation's most powerful supercomputers, teams of researchers are pushing the limits of what we know about HIV and how we can treat it.
Below are nine examples of how scientists are applying massive computing power and computational know-how to combat the disease.
1) Modeling HIV: from atoms to actions

An atomic-resolution simulation of the structure of the HIV capsid (credit: Theoretical and Computational Biophysics Group, University of Illinois Urbana-Champaign).
In order for HIV to infect non-dividing cells, the HIV virus must enter the cell and entice cellular proteins to act as chaperones, ushering the virus toward the cell nucleus and helping it integrate its genes into the cell's genome. This infection process offers opportunities for medical intervention and may suggest new HIV treatments. However the dynamics of the process can only be "observed" through computational modeling and simulation.
The size of the HIV capsid, or shell, combined with its irregular shape, had long prevented scientists from simulating the full capsid structure with adequate resolution. But researchers from Klaus Schulten's group at the University of Illinois at Urbana-Champaign, using the National Science Foundation-funded Blue Waters supercomputer, observed how the capsid interacts with drugs and host proteins at the atomic level. The model, consisting of about 1,300 proteins and 4 million atoms, is currently the largest entry in the Research Collaboratory for Structural Bioinformatics Protein Data Bank, a repository for the three-dimensional structural data of large biological molecules.
2) Discovery of hidden pocket in HIV protein leads to ideas for new inhibitors
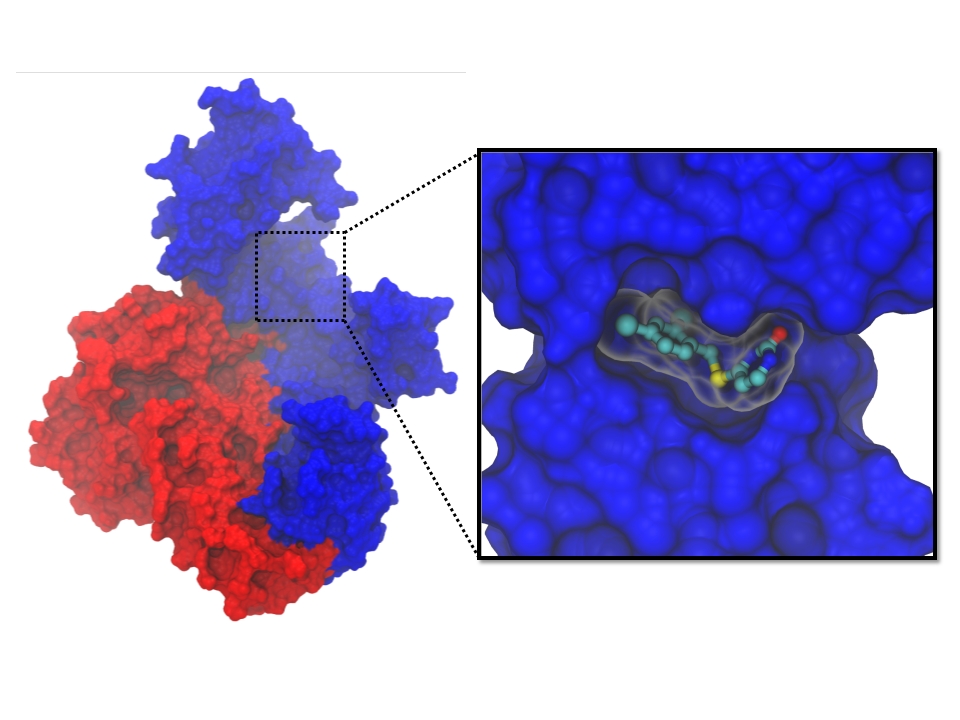
A hidden pocket within a vital HIV protein can be targeted by drugs that act as "molecular wedges" (credit: Anthony Ivetac, Sara E. Swift and J. Andrew McCammon, Department of Chemistry and Biochemistry, University of California at San Diego; John Naughton, Arturo Diaz and John A. T. Young, Nomis Center for Immunobiology and Microbial Pathogenesis, Salk Institute for Biological Studies; Paul L. Boyer and Stephen H. Hughes, HIV Drug Resistance Program, Center for Cancer Research, National Cancer Institute).
Researchers from the University of California San Diego, the Salk Institute for Biological Studies and the National Cancer Institute collaborated on an effort to discover new drug candidates to combat HIV.
With the help of the San Diego Supercomputer Center, the scientists ran molecular simulations to capture the movements of a small pocket on the virus' surface that they believed could be targeted by drugs to prevent the replication of the virus. Using the pocket as a target, they virtually screened thousands of compounds and tested 16 for their ability to block HIV infection in human tissue cultures. Ultimately, they discovered two compounds that inhibit HIV replication and block the activity of reverse transcriptase as effectively as a leading FDA-approved drug, nevirapine. The researchers believe these compounds have the potential to develop into future drugs and are exploring them further.
3) Preventing HIV from reaching its mature state

Coarse-grained molecular model of a mature HIV-1 viral capsid shows its armor-like structure (credit: Gregory A. Voth, University of Chicago).
The mature capsid of the HIV virus comprises thousands of interlinked proteins that act like a suit of armor around the virus' genetic material. If this armor-like structure does not form, then the virus is unable to infect cells.
Researchers from the University of Chicago used the Kraken supercomputers at the National Institution for Computational Science (NICS) to study how the mature HIV capsid formed. They found that the seemingly complicated behavior of the capsid's self-assembly was relatively simple once they understood the shape and behavior of the proteins that made it up. The work advanced our understanding of the HIV life cycle and is inspiring the development of new drugs to disrupt the virus' growth. Results appeared in the Biophysical Journal in October 2012.
4) Crowdsourcing a cure
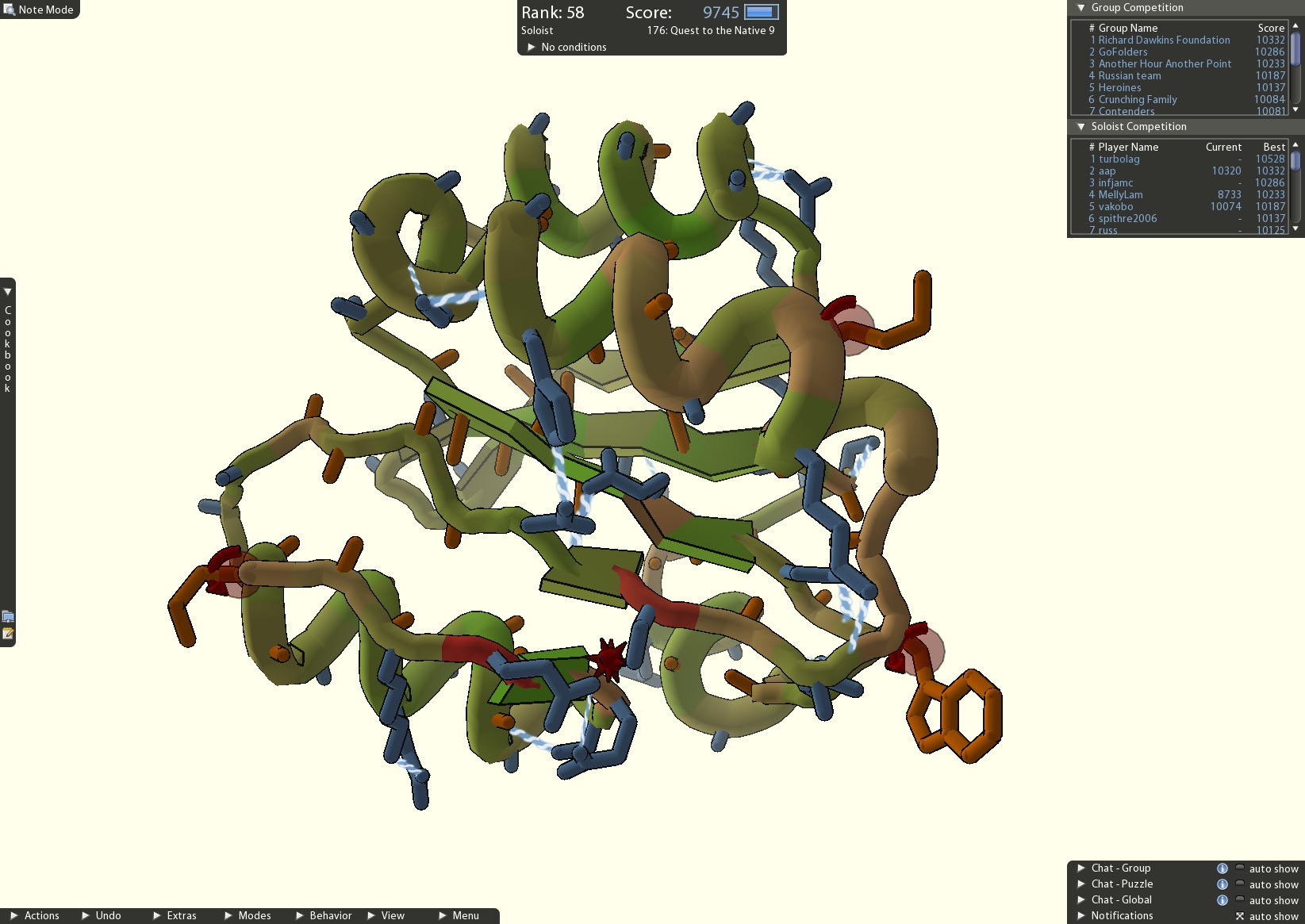
A screenshot of an active Foldit puzzle; the leaderboard shows the top score of each player/team (credit: University of Washington).
After scientists repeatedly failed to piece together the structure of a protein-cutting enzyme that plays an important role in HIV, they called on the players of FoldIt, an online puzzle video game, to find a solution. Using FoldIt "citizen scientists" were able to determine how the enzyme folded and solved the mystery of its structure. With further help from the game players, researchers were able to identify target drugs to neutralize the enzyme.
FoldIt is part of an experimental research project supported by NSF and developed by the University of Washington's Center for Game Science in collaboration with the UW Department of Biochemistry. The case of the crowdsourced protein structure serves as a critical example of how games with a purpose can solve real-world problems.
5) Virtual screening of HIV inhibitors
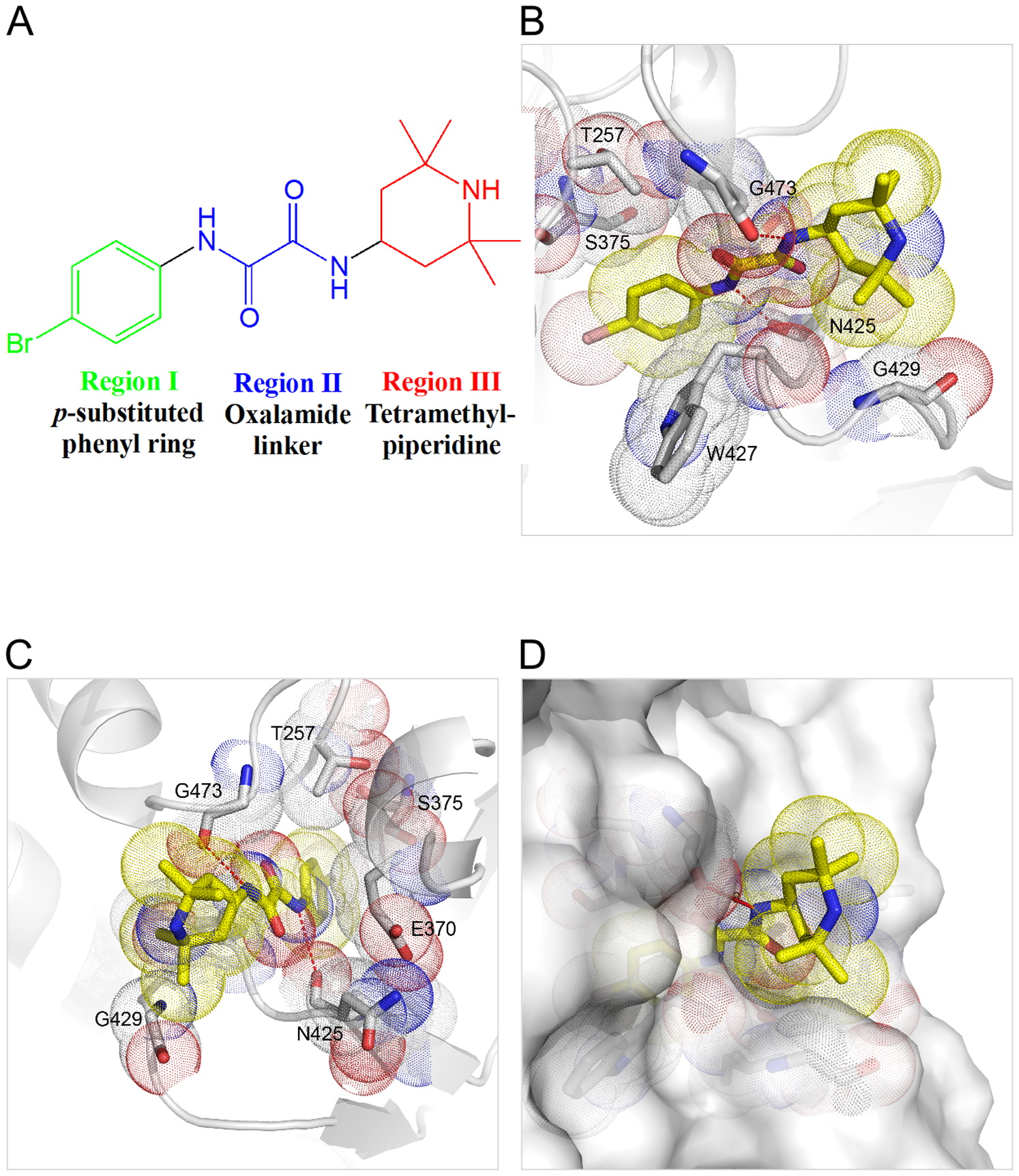
Scientists simulated two proteins that play a key role in HIV in order to find novel inhibitors (credit: Kwon YD, LaLonde JM, Yang Y, Elban MA, et al.).
A team of researchers from Pennsylvania used computer modeling and virtual screening, powered by supercomputers, to identify novel inhibitors of HIV and better understand how they react with the HIV virus. They focused on small molecules that block the interaction between the receptors on the surface of human cells and an important protein on the surface of the HIV envelope.
Using the Blacklight system at the Pittsburgh Supercomputing Center, the researchers virtually screened more than 10 million compounds to find small molecules that would be a good molecular fit for the protein that they were targeting. From the 10 million they identified six small-molecule, HIV-surface protein complexes that display unique modes of binding. Taken together they constitute what the researchers believe is a potent class of entry inhibitors against HIV.
6) Membrane effects
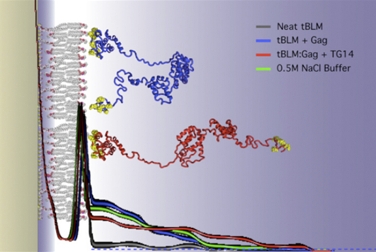
The simulated structure of the HIV-1 viral assembly protein Gag (credit: Hirsh Nanda, National Institute of Standards and Technology).
Some proteins that anchor HIV to cell membranes are thought to promote the development of the virus. Researchers have found that combining experimental methods with computer simulations can reveal much about the cell-binding dynamics.
Hirsh Nanda of the National Institute of Standards and Technology leads a research team that studies the initial stages of the formation of new HIV virus particles in an infected cell. During these first steps, HIV proteins latch onto cell membranes.
Using the Kraken supercomputer at the National Institute for Computational Science, Nanda's team was able to study the forces that govern protein assemblies on membranes in far greater detail and much faster than if they were using their lab's computers. Kraken also greatly accelerated the analysis of experimental neutron-scattering data that they used to compare with simulations.
The simulations revealed that an important HIV surface protein simultaneously binds to the cell membrane and to viral RNA in order to change shape, and how another HIV protein transitions between compact and extended structures upon anchoring to the cell membrane. These discoveries are inspiring new treatment approaches that center on membrane interactions.
7) Computing patient-specific treatment methods
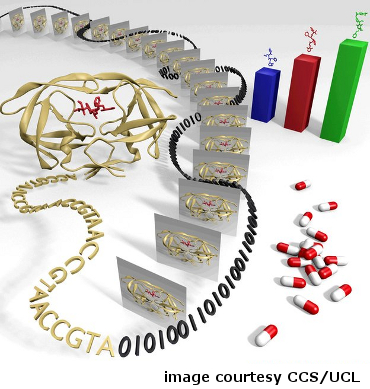
Scientists translate HIV sequence data into digitized information to rank HIV inhibitors (credit: Hugh Martin, Dave Wright and Peter Coveney, Centre for Computational Science, University College London).
Doctors know that there are many different strains of HIV, and that drugs for the disease do not have the same effects in all people. Subtle genetic differences between strains and among individuals lead to a range of treatment outcomes. Using the NSF-supported Kraken and Ranger supercomputers, researchers from University College London and Rutgers University determined the shape of a key protein involved in HIV infection in an individual patient and then ranked the drug molecules most likely to block the activity.
The project demonstrated how researchers might use genetic-sequencing techniques and massive computations to design patient-specific treatment protocols in near-real time. In the future it is expected that this type of patient-specific drug selection will become routine.
The research was reported at the annual meeting of the American Association for the Advancement of Science and was published in the Journal of Chemical Theory and Computation.
8) Preparing the next generation to continue the fight

Merrimack College undergraduates Daniel Laverty (back) and Dave Daniels (front) evaluating the hits from the virtual screening using PyMOL (credit: Jimmy Franco, Merrimack College).
At Merrimack College in Massachusetts, students are learning how to conduct virtual screening using the Stampede supercomputer at the Texas Advanced Computing Center. Virtual screening uses computational methods to identify small molecules that are likely to bind to a known drug target, often a protein. The method has become a valuable tool for many biotechnology and pharmaceutical companies.
The activity exposes students to massive computing resources and shows them a method of conducting science that few previously knew existed. It's one of many ways that educators around the nation are beginning to prepare students for the workforce of the future by incorporating computational techniques into their curriculum.
9) A boy and the BEAST

Armand Bilge next to a poster explaining his award-winning HIV research project (credit: Armand Bilge, Lexington High School).
When Armand Bilge was a 10th grader at Lexington High School in Massachusetts, he created a map and timeline that identified when HIV arrived in the Americas, and where and when HIV spread across these continents. To do so, Bilge used a combination of molecular-sequencing software and NSF-funded high-performance computing resources.
As a member of an after-school computer club, Bilge used a software program called BEAST to create a detailed evolutionary tree based on similarities and differences in the 3,000 nucleotide subunits of a gene among 400 known HIV strains. The software ran on the CIPRES (CyberInfrastructure for Phylogenetic Research) science gateway, a public resource developed by the San Diego Supercomputer Center and supported by NSF that allows those interested in evolutionary relationships to study virtually every species on Earth.
Bilge's conclusions support previously published results of HIV experts that suggest that "a single introduction of the virus in Haiti in the mid-1900s resulted in its dispersion across the American continent." The project won first place in biology for the 2012 Massachusetts Science and Engineering Fair.